
OSILODROSTAT 10 mg, comprimé pelliculé, boîte de 1 flacon de 40
Retiré du marché le : 02/06/2020
Dernière révision : 15/01/2020
Taux de TVA : 0%
Laboratoire exploitant : NOVARTIS PHARMA
Source :

Osilodrostat est indiqué dans le traitement du syndrome de Cushing endogène chez l'adulte.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Hypocorticisme
L'inihibition de la synthèse du cortisol par Osilodrostat a entraîné des événements liés à un hypocorticisme tels qu'un syndrôme de sevrage du cortisol (diminution symptomatique des concentrations de cortisol, mais toujours au-dessus de la limite inférieure de l'intervalle normal) et une insuffisance surrénalienne (concentrations de cortisol inférieures à l'intervalle normal).
Les concentrations de cortisol doivent être surveillées à intervalles réguliers car les événements liés à un hypocorticisme peuvent survenir à tout moment pendant le traitement, en particulier lorsque les besoins en cortisol sont accrus, notamment lors d'un stress physique ou psychologique (voir rubrique Posologie et mode d'administration). Il est recommandé d'utiliser des méthodes biologiques ne présentant pas de réactivité croisée significative avec les précurseurs du cortisol tels que le 11 désoxycortisol, susceptibles d'augmenter au cours du traitement par osilodrostat.
Les patients doivent être informés des signes et symptômes associés à l'hypocorticisme (ex : nausées, vomissements, fatigue, douleurs abdominales, perte d'appétit et étourdissements). Il est également conseillé aux patients de découper et de porter sur eux la Carte Patient indiquant qu'ils courent un risque d'hypocorticisme et que des mesures appropriées doivent être prises si des soins d'urgence sont nécessaires.
Chez les patients présentant des symptômes, il convient de surveiller l'hypotension, l'hyponatrémie, l'hyperkaliémie et/ou l'hypoglycémie. En cas de suspicion d'hypocorticisme, les concentrations de cortisol doivent être mesurées et une réduction ou une interruption temporaire d'Osilodrostat doit être envisagée. Si nécessaire, un traitement de substitution par corticostéroïdes doit être instauré.
Osilodrostat pourra être repris à une dose inférieure après résolution des symptômes, à condition que les concentrations de cortisol soient supérieures à la limite inférieure de la normale en l'absence de traitement de substitution par glucocorticoïdes.
Allongement de l'intervalle QT
Dans une étude approfondie de l'intervalle QT, Osilodrostat a été associé à un allongement dose- dépendant de l'intervalle QT, (allongement maximal de l'intervalle QTcF estimé en moyenne à +5,3 ms à la dose maximale recommandée de 30 mg) qui peut être responsable d'arythmies cardiaques (voir rubrique Propriétés pharmacodynamiques). Un ECG doit être réalisé avant le début du traitement par Osilodrostat, dans la semaine qui suit l'instauration du traitement et ensuite si cliniquement indiqué. Si l'intervalle QTc dépasse 480 ms avant ou pendant le traitement, il est recommandé d'effectuer une consultation cardiologique.
Toute hypokaliémie, hypocalcémie ou hypomagnésémie doit être corrigée avant l'administration d'Osilodrostat et leurs concentrations doivent être surveillées périodiquement pendant le traitement.
Chez les patients présentant des facteurs de risque d'allongement de l'intervalle QT (comme le syndrome congénital du QT long, l'insuffisance cardiaque congestive, les bradyarythmies, les anomalies électrolytiques non corrigées et la prise concomitante de médicaments connus pour allonger l'intervalle QT) et chez les patients avec une pathologie cardiovasculaire significative, il conviendra d'être prudent et une surveillance par ECG plus fréquente est recommandée.
Femmes en âge de procréer
Osilodrostat peut être nocif pour le foetus. L'absence de grossesse devra être vérifiée chez les femmes en âge de procréer avant l'instauration d'Osilodrostat, et ces patientes doivent être informées du risque potentiel pour le foetus et de la nécessité d'utiliser une contraception efficace pendant le traitement et pendant au moins une semaine après l'arrêt du traitement (voir rubrique Fertilité, grossesse et allaitement).
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés dans l'étude pivot de phase III avec Osilodrostat ont été l'insuffisance surrénale (51%), fatigue (44%), oedème (21%), vomissements (22%), nausées (42%) et céphalées (34%).
L'effet indésirable le plus grave associé à l'utilisation d'Osilodrostat est l'insuffisance surrénalienne (voir aussi les rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi).
Les effets indésirables (Tableau 1) sont présentés par classe de systèmes d'organes MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés par ordre de fréquence décroissante. Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante. De plus, la catégorie de fréquence correspondant à chaque effet indésirable médicamenteux est définie selon la convention suivante (CIOMS III) : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000).
Tableau 1 Evénements indésirables
| Classe de systèmes d'organes | Catégorie de fréquence | Terme préférentiel |
| Affections endocriniennes | Très fréquent | Insuffisance surrénale* |
| Troubles du métabolisme et de la nutrition | Très fréquent | Hypokaliémie*, perte d'appêtit |
| Affections du système nerveux | Très fréquent | Etourdissements*, céphalée* |
| Fréquent | Syncope* | |
| Affections cardiaques | Fréquent | Tachycardie* |
| Affections vasculaires | Très fréquent | Hypotension* |
| Affections gastro-intestinales | Très fréquent | Vomissements, nausée, diarrhée, douleurs abdominales* |
| Affections de la peau et du tissu sous cutané | Très fréquent | Rash* |
| Fréquent | Hirsutisme*,**, acné** | |
| Troubles généraux et anomalies au site d'administration | Très fréquent | Fatigue*, oedème* |
| Fréquent | Malaise | |
| Investigations | Très fréquent | Augmentation du taux de testostérone**, augmentation du taux de corticotrophine (ACTH) |
| Fréquent | Allongement du QT à l'électrocardiogramme, Augmentation des transaminases* | |
| * Correspond au regroupement de deux termes préférentiels MedDRA ou plus qui ont été jugés similaires sur le plan clinique. Le terme « insuffisance surrénale » comprend les termes carence en glucocorticoïdes, insuffisance cortico-surrénale aiguë, syndrome de sevrage des stéroïdes, diminution de l'urine sans cortisol, diminution du cortisol. ** Catégorie de fréquence « très fréquent » chez les patientes. | ||
Description des effets indésirables sélectionnés
L'inhibition du CYP11B1 par l'osilodrostat est associée à une accumulation de précurseurs corticostéroïdes et à une augmentation de la testostérone. Dans une étude clinique avec l'osilodrostat, les taux moyens de testostérone chez les patientes ont augmenté, passant d'abord d'une normale élevée à la limite supérieure de la normale. Ces tendances se sont inversées lorsque le traitement a été interrompu. L'augmentation de la testostérone était associée à des cas légers à modérés d'hirsutisme ou d'acné chez un sous-groupe de patients.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable au moyen de la fiche correspondante du protocole d'utilisation thérapeutique (voir annexe B4).
SURVEILLANCE
du traitement :
- Réaliser
un test de grossesse avant de débuter le traitement.
- Réaliser
un ECG avant le début du traitement, puis dans la semaine qui suit
l'instauration du traitement et ensuite si cliniquement indiqué.
- Dosage
du cortisol (ex : cortisol libre dans les urines des 24 heures, cortisol
sérique/plasmatique) chaque semaine ou toutes les deux semaines jusqu'au
maintien d'une réponse clinique stable. Ensuite, selon l'état clinique, une
surveillance moins fréquente pourra être envisagée.
Il est
recommandé d'utiliser des méthodes biologiques ne présentant pas de réactivité
croisée significative avec les précurseurs du cortisol tels que le 11
désoxycortisol, susceptibles d'augmenter au cours du traitement par
osilodrostat.
-
Dosage périodique de la kaliémie, de la calcémie et de la magnésémie.
INFORMER
immédiatement un MEDECIN en cas de
- Trouble
cardiaque ou un trouble du rythme cardiaque, comme des battements irréguliers.
- Faiblesse,
étourdissements, fatigue (épuisement), manque d'appétit, envie de vomir,
vomissements.
CONTRACEPTION :
les femmes en âge de procréer doivent utiliser une méthode de contraception
efficace pendant le traitement et jusqu'à au moins une semaine après l'arrêt du
traitement.
ALLAITEMENT :
ne pas allaiter au cours du traitement et pendant au moins une semaine après arrêt du traitement.
PRUDENCE en cas de conduite de véhicule ou d'utilisation de
machines (vertige, fatigue).
Grossesse
Les données concernant l'utilisation de l'osilodrostat chez la femme enceinte sont inexistantes ou limitées. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité précliniques). Osilodrostat n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement
On ne sait pas si osilodrostat est excrété dans le lait maternel. Un risque pour les nouveau- nés/nourrissons ne peut être exclu. L'allaitement doit être interrompu au cours du traitement par Osilodrostat et pendant au moins une semaine après arrêt du traitement.
Fertilité
Aucune information n'est disponible concernant l'effet de l'osilodrostat sur la fertilité humaine. Des études sur l'animal ont montré des effets sur le cycle menstruel et une réduction de la fertilité des rats femelles à des doses élevées (voir rubrique Données de sécurité précliniques).
Effets d'autres médicaments sur le métabolisme de l'osilodrostat
L'osilodrostat a montré une perméabilité intrinsèque élevée, un faible taux d'efflux et un impact modéré des inhibiteurs sur le taux d'efflux in vitro. De plus, plusieurs enzymes du CYP et des UDP- glucuronosyltransférases contribuent au métabolisme de l'osilodrostat, et aucune enzyme ne contribue seule à plus de 25 % de la clairance totale. Ceci suggère que le potentiel d'interactions médicamenteuses cliniques (DDI) avec des médicaments administrés de manière concomitante qui inhibent les transporteurs ou une seule enzyme du CYP ou de l'UGT est faible. Toutefois, il conviendra d'être prudent lorsque de puissants inhibiteurs du CYP ou de puissants inducteurs d'enzymes non spécifiques (ex : la rifampicine, le phénobarbital) sont introduits ou arrêtés lors du traitement par osilodrostat.
Effets de l'osilodrostat sur le métabolisme d'autres médicaments
Dans une étude incluant des volontaires sains (n=20) utilisant une dose unique de 50 mg d'osilodrostat associés à plusieurs autres médicaments, l'osilodrostat s'est révélé être un faible inhibiteur de CYP2D6 et CYP3A4/5, un inhibiteur faible à modéré de CYP2C19 et un inhibiteur modéré de CYP1A2. L'osilodrostat doit être utilisé avec précaution lorsqu'il est administré en association avec des substrats de CYP2C19 et de CYP1A2 qui ont un index thérapeutique étroit, comme la théophylline, la tizanidine et la S-méphénytoïne.
Dans une étude incluant des volontaires sains (n=24), l'osilodrostat (30 mg deux fois par jour pendant 12 jours) n'a pas eu d'effet clinique significatif sur les concentrations plasmatiques d'un contraceptif oral combiné contenant 0,03 mg d'éthinylestradiol et 0,15 mg de lévonorgestrel. Les ratios des moyennes géométriques de l'ASC ont été de 1,03 pour l'éthinylestradiol et de 1,02 pour le lévonorgestrel. D'après cette étude, aucune interaction pharmacocinétique n'est attendue avec les contraceptifs hormonaux.
Interactions pharmacodynamiques potentielles
L'administration concomitante d'osilodrostat avec d'autres traitements connus pour affecter l'intervalle QT peut entraîner un allongement de l'intervalle QT chez les patients présentant des troubles du rythme cardiaque connus (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacodynamiques).
Le traitement doit être initié et supervisé par des médecins expérimentés en endocrinologie ou en médecine interne et qui ont les compétences appropriées pour le suivi des réponses biochimiques afin que la dose soit ajustée pour atteindre les besoins thérapeutiques du patient, en se basant sur la normalisation des concentrations de cortisol.
Posologie
La dose recommandée d'Osilodrostat est de 2 mg deux fois par jour. Pour les patients d'origine asiatique, une réduction de la dose initiale de 1 mg deux fois par jour est recommandée (voir rubrique Propriétés pharmacocinétiques).
La dose peut être progressivement augmentée (initialement par paliers de 1 ou 2 mg deux fois par jour) en fonction de la réponse individuelle et de la tolérance, afin d'atteindre des concentrations normales de cortisol. Il est recommandé de surveiller les concentrations de cortisol (ex : cortisol libre dans les urines des 24 heures, cortisol sérique/plasmatique) chaque semaine ou toutes les deux semaines jusqu'au maintien d'une réponse clinique stable. Ensuite, selon l'état clinique, une surveillance moins fréquente pourra être envisagée.
La dose d'Osilodrostat doit être diminuée ou le traitement doit être provisoirement interrompu si les concentrations de cortisol sont inférieures à la limite inférieure de la normale, ou si l'on observe une diminution rapide des concentrations de cortisol vers la limite inférieure de la plage normale, ou si le patient présente des signes ou symptômes suggérant un hypocorticisme (voir rubrique Mises en garde et précautions d'emploi). La prise en charge d'autres effets indésirables à tout moment du traitement peut également nécessiter une réduction de dose ou une interruption temporaire du traitement.
La dose d'entretien habituelle dans les essais cliniques était comprise entre 2 et 7 mg deux fois par jour.
La dose maximale recommandée d'Osilodrostat est de 30 mg deux fois par jour.
En cas d'oubli d'une dose, le patient doit prendre la dose prescrite lors de la prise suivante ; la dose suivante ne doit pas être doublée.
Personnes âgées (65 ans ou plus)
Les données sur l'utilisation de l'osilodrostat chez les patients de plus de 65 ans sont limitées, mais il n'existe aucun argument suggérant qu'une adaptation de la posologie soit nécessaire chez ces patients.
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance rénale (voir rubrique Propriétés pharmacocinétiques). Les concentrations de cortisol libre urinaire (CLU) doivent être interprétées avec prudence chez les patients présentant une insuffisance rénale modérée à sévère, en raison de l'excrétion réduite de CLU. Des méthodes alternatives de surveillance du cortisol doivent être envisagées chez ces patients.
Insuffisance hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance hépatique légère (Child-Pugh A). Pour les patients présentant une insuffisance hépatique modérée (Child- Pugh B), la dose initiale recommandée est de 1 mg deux fois par jour. Pour les patients présentant une insuffisance hépatique sévère (Child-Pugh C), la dose initiale recommandée est de 1 mg une fois par jour, le soir, avec une augmentation de dose initiale de 1 mg deux fois par jour (voir rubrique Propriétés pharmacocinétiques).
Une surveillance plus fréquente de la fonction surrénale peut être nécessaire chez les patients présentant une insuffisance hépatique pendant la période de titration de la dose.
Population pédiatrique
La sécurité et l'efficacité d'Osilodrostat chez les enfants âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie orale.
Osilodrostat peut être pris avec ou sans nourriture.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite aucune conditions de conservation particulière de température. A conserver dans l'emballage extérieur d'origine à l'abri de l'humidité.
Sans objet
Un surdosage peut entraîner un hypocorticisme sévère. Les signes et symptômes évocateurs d'un hypocorticisme peuvent inclure des nausées, vomissements, fatigue, hypotension, douleur abdominale, perte d'appétit, vertige et syncope.
En cas de suspicion de surdosage, Osilodrostat doit être interrompu, les concentrations de cortisol doivent être vérifiés et, si nécessaire, une supplémentation en corticostéroïdes doit être instaurée. Une surveillance étroite peut être nécessaire, incluant la surveillance de l'intervalle QT, de la tension artérielle, du glucose, et de l'équilibre hydro-électrolytique, jusqu'à ce que l'état du patient soit stable.
Classe pharmacothérapeutique : non encore attribuée. Code ATC : non encore attribué
Mécanisme d'action
L'osilodrostat est un inhibiteur de la synthèse du cortisol. Il inhibe puissamment la 11β-hydroxylase (CYP11B1), enzyme responsable de l'étape finale de biosynthèse du cortisol dans la glande surrénale.
Dans une analyse in vitro, l'osilodrostat a inhibé, de façon dose-dépendante, l'activité de la 11β- hydroxylase recombinante humaine avec une CI50 de 2,5 nM.
L'inhibition du CYP11B1 est associée à l'accumulation de précurseurs tels que le 11-désoxycortisol et à l'accélération de la biosynthèse des glandes surrénales, y compris des androgènes. Dans la maladie de Cushing, la baisse de la concentration plasmatique de cortisol stimule également la sécrétion d'ACTH via le mécanisme de rétroaction qui accélère la biosynthèse des stéroïdes.
Effets pharmacodynamiques
Dans une étude approfondie de l'intervalle QT (n=86 hommes et femmes, volontaires sains) avec l'osilodrostat, les différences maximales de durée de l'intervalle QTcF comparativement au placebo étaient de 1,73 ms (IC à 90% : 0,15 ;3,31) à la dose de 10 mg et de 25,38 ms (IC à 90 % : 23,53 ; 27,22) à une dose supra-thérapeutique de 150 mg. Sur la base d'une extrapolation de ces résultats, l'allongement maximal moyen à la dose maximale recommandée de 30 mg devrait être de +5,3 ms.
Efficacité et sécurité cliniques
L'efficacité et la sécurité de l'osilodrostat chez les patients atteints de la maladie de Cushing ont été évaluées dans une étude prospective de phase III (étude C2301) utilisant un schéma d'arrêt randomisé. L'étude consistait en une période ouverte de 26 semaines de traitement par osilodrostat dans un bras, suivie d'une période d'arrêt randomisée de 8 semaines au cours de laquelle les patients étaient randomisés selon un rapport de 1:1 dans un bras osilodrostat ou un bras placebo suivi d'une période de traitement par osilodrostat en ouvert.
Les critères d'éligibilité comprenaient la maladie de Cushing (avec confirmation de la source hypophysaire d'hormone de l'axe adrénocorticotrope en excès), et une valeur moyenne de cortisol libre urinaire (CLUm, dérivé de trois collectes d'urine de 24 heures) supérieure à 1,5 fois la limite supérieure de la normale (LSN) au moment du dépistage.
Au total, 137 patients adultes ont été inclus. L'âge moyen était de 41,2 ans et la majorité des patients étaient des femmes (77%). Sept patients étaient âgés de 65 ans ou plus. Le traitement antérieur comprenait une chirurgie hypophysaire chez 88% des patients et un traitement médical antérieur chez 75% des patients. Les concentrations moyennes et médianes initiales de CLUm étaient respectivement de 1006,0 nmol/24h et de 476,4 nmol/24h (LSN : 138 nmol/24h). Au début de l'essai, les comorbidités comprenaient l'hypertension (67,9% des patients), l'obésité (29,9%), le diabète de type II (21,9%) et l'ostéoporose (27,7%).
Les patients ont reçu une dose initiale de 2 mg d'osilodrostat deux fois par jour et la dose pouvait être augmentée en fonction de la réponse individuelle et de la tolérance durant une période initiale de 12 semaines. Les patients sans autre augmentation de dose au cours des 12 semaines suivantes et avec une CLUm ≤LSN à la semaine 24 ont été randomisés selon un ratio de 1:1 à la semaine 26 pour recevoir soit osilodrostat, soit un placebo pendant 8 semaines (période d'arrêt randomisée en double aveugle), suivie par une administration d'osilodrostat en ouvert pour le reste de l'étude. À la semaine 26, 71 patients ont été randomisés selon un ratio de 1:1 pour continuer à recevoir l'osilodrostat (n=36) ou pour passer au placebo (n=35). Les patients qui n'étaient pas éligibles à la randomisation à la semaine 24 (n=47) ont poursuivi le traitement par osilodrostat en ouvert.
L'objectif principal était de comparer la proportion de répondeurs complets à la semaine 34 (fin de la période de 8 semaines d'arrêt randomisé) entre les patients randomisés poursuivant le traitement actif et le placebo. Pour le critère principal, une réponse complète a été définie comme une valeur de CLUm ≤LSN à la semaine 34. Les patients dont la dose a été augmentée au cours de la période de sevrage randomisé ou qui ont arrêté le traitement randomisé ont été considérés comme non- répondeurs. Le critère principal secondaire était le taux de réponse complète à la semaine 24. Les patients ayant reçu une augmentation de la dose entre les semaines 12 et 24 et les patients sans évaluation valide du CLUm à la semaine 24 ont été comptés comme non répondeurs pour le critère principal secondaire.
L'étude a atteint ses critères principaux primaires et secondaires (Tableau 2). À la semaine 34, la réponse était maintenue chez une proportion significativement plus élevée de patients du groupe ayant poursuivi le traitement actif (86,1%) par rapport à ceux qui étaient passés au placebo (29,4%).
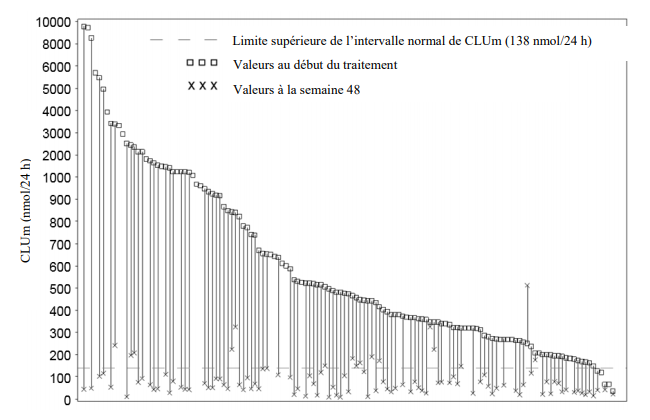
Les concentrations médianes de CLUm ont diminué à 62,5 nmol/24h à la 12e semaine (variation de 84,1% par rapport au début, n=125), à 75,5 nmol/24h à la semaine 24 (82,3%, n=125) ;et 63,3 nmol/24h à la semaine 48 (87,9%, n=108). Les changements individuels entre le commencement de l'étude et la semaine 48 sont présentés dans la Figure 1.
Tableau 2 Résultats principaux : Etude de phase III chez les patients atteints de la maladie de Cushing (étude C2301)
| | Osilodrostat n=36 | Placebo n=34 | |
| Critère primaire : Proportion de répondeurs à la fin de la période d'arrêt randomisé (semaine 34) n (%) (IC à 95%) | 31 (86,1) (70,5 ; 95,3) | 10 (29,4) (15,1 ; 47,5) | |
| Différence de taux de réponse (odds ratio) : osilodrostat vs. placebo | 13,7 (3,7; 53,4) 2-sided p value <0,001 | | |
| Critères secondaires | Tous les patients N=137 | ||
| Critère secondaire clé : proportion avec CLUm ≤LSN à la semaine 24 et sans augmentation de dose après la semaine 12 (IC à 95%) | 72 (52,6%) (43,9 ; 61,1) | ||
| Taux de réponse complète de CLUm (CLUm ≤LSN) à la semaine 48 | 91 (66,4%) (57,9 ; 74,3) | ||
| Valeur médiane du CLUm et différence de pourcentage à la semaine 48 | 63,3 nmol/24 h (-87,9%) | ||
| CLUm : cortisol libre urinaire moyen ; LSN : limite supérieure de la normale ; IC : intervalle de confiance ; réponse : CLUm ≤LSN. | |||
Figure 1 Valeurs individuelles du CLUm par patient au début du traitement et à la semaine 48 (étude C2301)
Des améliorations des paramètres cardiovasculaires et métaboliques ont été observées (Tableau 3) et 85,6% des patients dont l'évaluation était disponible ont montré une amélioration d'au moins une caractéristique physique de la maladie de Cushing à la semaine 48.
Table 3 Paramètres cardiovasculaires et métaboliques
| | Début de traitement | Semaine 24 | Semaine 48 |
| Pression artérielle systolique (mmHg) | 132,2 | 124,9 (-4,1%) | 121,7 (-6,8%) |
| Pression artérielle diastolique (mmHg) | 85,3 | 81,0 (-3,8%) | 78,9 (-6,6%) |
| Poids (kg) | 80,8 | 77,3 (-3,0%) | 75,5 (-4,6%) |
| Tour de taille (cm) | 103,4 | 99,1 (-2,6%) | 97,4 (-4,2%) |
| HbA1c (%) | 6,0 | 5,6 (-4,6%) | 5,6 (-5,4%) |
Le traitement par osilodrostat a également entraîné une amélioration des résultats rapportés par les patients. Des améliorations supérieures à la différence importante minimale établie (DIM) ont été observées sur la qualité de vie des patients atteints de syndrome de Cushing (score total, sous- échelle des Problèmes Physiques et sous-échelle des Problèmes Psychosociaux), sur l'indice d'utilité EQ 5D et sur les scores BDI II (dépression). Le score total moyen de qualité de vie du Cushing est passé de 42,2 au départ 58,3 (+14,1 ; variation de + 52,4% par rapport au départ) à la 48e semaine.
L'efficacité de l'osilodrostat a également été évaluée dans le cadre de l'étude C1201 chez des patients adultes japonais présentant des causes non hypophysaires du syndrome de Cushing. L'étude incluait des patients atteints d'un adénome surrénalien (n=5), d'un syndrome de Cushing par sécrétion ectopique d'ACTH (n=3) et d'une hyperplasie surrénale macronodulaire indépendante de l'ACTH (n=1), et consistait en une période de titration de la dose de 12 semaines (dose initiale 2 mg deux fois par jour), une période de maintenance de 36 semaines et une extension à long terme optionnelle. À la 12e semaine (critère primaire), une réponse complète (CLUm ≤ LSN) a été observée chez 6 patients (66,7%) et une réponse partielle (diminution du CLUm d'au moins 50%) chez un patient supplémentaire (11,1%). La dose moyenne médiane utilisée dans l'étude était de 2,6 mg/jour (intervalle de 1,3 à 7,5 mg/jour).
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Osilodrostat dans un ou plusieurs sous-groupes de la population pédiatrique dans l'hyperfonction cortico-surrénale (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
L'osilodrostat est un composé hautement soluble et hautement perméable (BCS de classe 1). Il est rapidement absorbé (Tmax ~ 1 h) et l'absorption orale chez l'homme est supposée quasiment complète. L'état d'équilibre est atteint au Jour 2.
La co-administration avec des aliments n'a pas eu d'incidence significative sur l'absorption. Dans une étude sur des volontaires sains (n=20), l'administration d'une dose unique de 30 mg d'osilodrostat avec un repas riche en graisse a entraîné une réduction modérée de l'ASC et de la Cmax de 11% et 21%, respectivement, et le Tmax médian a été retardé de 1 à 2,5 heures.
Aucune accumulation cliniquement pertinente ni auto-induction n'a été observée dans les études cliniques.
Distribution
Le volume de distribution apparent médian (Vz/F) de l'osilodrostat est d'environ 100 litres. La liaison aux protéines est faible (36,4%) et indépendante de la concentration. Le rapport entre la concentration sanguine et la concentration plasmatique de l'osilodrostat est de 0,85.
Biotransformation
Dans une étude ADME chez des sujets sains humains après l'administration d'une dose unique de 50 mg d'osilodrostat-[14C], le métabolisme a été considéré comme la principale voie de clairance de l'osilodrostat car environ 80% de la dose était excrétée sous forme de métabolites. Les enzymes de plusieurs CYP et les UDP-glucuronosyltransférases contribuent au métabolisme de l'osilodrostat et aucune enzyme seule ne contribue à plus de 25% de la clairance totale. Les métabolites ne sont pas censés contribuer à l'effet pharmacologique de l'osilodrostat.
Elimination
Dans une étude ADME, la majeure partie (90,6%) de la dose radioactive d'osilodrostat était éliminée dans les urines, et une quantité minime dans les fèces (1,6% de la dose). Le faible pourcentage de la dose éliminée dans l'urine sous la forme d'osilodrostat inchangé (5,2%) indique que le métabolisme est la principale voie de clairance chez l'homme.
La demi-vie d'élimination de l'osilodrostat est d'environ 4 heures.
Linéarité/non-linéarité
L'exposition (ASCinf etCmax) a augmenté de manière sur-proportionnelle par rapport à la dose au-delà de l'intervalle de doses thérapeutiques.
Populations spéciales
L'âge et le sexe n'ont eu aucun impact significatif sur l'exposition à l'osilodrostat chez l'adulte.
Race/ethnie
La biodisponibilité relative était environ 20% plus élevée chez les patients asiatiques que chez les autres groupes ethniques. Il n'a pas été démontré que le poids était un facteur déterminant de cette différence.
Insuffisance rénale
Dans une étude de phase I chez 15 sujets présentant différents degrés de fonction rénale utilisant une dose unique de 30 mg d'osilodrostat, la Cmax était légèrement inférieure chez les sujets présentant une insuffisance rénale grave (DFG absolu calculé ≥15 et <29 ml/minute) et chez les sujets présentant une IRT (DFG absolu calculé <15 ml/minute ; les sujets souffrant d'IRT n'étaient pas sous dialyse) par rapport aux témoins (respectivement de 10 et 18%), avec une variation minime de l'ASC (1 à 4%). La fonction rénale n'influe pas de manière significative sur la pharmacocinétique de l'osilodrostat.
Insuffisance hépatique
Dans une étude de phase I chez 33 sujets présentant divers degrés de fonction hépatique, traités avec une dose unique de 30 mg d'osilodrostat, l'ASCinf était respectivement 1,4 et 2,7 fois plus élevée dans les cohortes d'insuffisance hépatique modérée (Child Pugh B) et sévère (Child Pugh C). La Cmax était 15 et 20% plus faible dans les cohortes modérées et sévères. La demi-vie terminale est passée à 9,3 heures et à 19,5 heures dans les cohortes modérées et sévères. Une insuffisance hépatique légère (Child Pugh A) n'a pas eu d'effet significatif sur l'exposition. Le taux d'absorption n'était pas affecté par le degré d'insuffisance hépatique. Un ajustement de la posologie initiale est recommandé en cas d'insuffisance hépatique modérée ou sévère (voir rubrique Posologie et mode d'administration).
Osilodrostat peut avoir une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Les patients doivent être prévenus du risque de vertiges et de fatigue (voir rubrique Effets indésirables) et il doit leur être recommandé de ne pas conduire ou utiliser de machines si ces symptômes apparaissent.
Des effets ont été observés chez l'animal uniquement à des expositions considérées comme suffisamment supérieures à l'exposition maximale observée chez l'homme, et ont peu de signification clinique
Pharmacologie de la sécurité
In vitro, l'osilodrostat inhibe les courants médiés par le canal hERG avec une CI50 au moins 80 fois supérieure à la Cmax humaine. Aucun effet sur les canaux Nav 1,5 et Cav 1,2 exprimés dans les cellules de mammifère n'a été observé jusqu'à 100 µM. Un potentiel arythmique a été observé dans une analyse cardiaque chez le lapin à une exposition égale à 15 fois l'exposition humaine (Cmax, libre) et une tachycardie ventriculaire non soutenue et des torsades de pointes ont été observées chez un singe présentant une exposition égale à 39 fois l'exposition humaine (Cmax, libre).
Toxicités à doses répétées
Dans les études de toxicité à doses répétées, le système nerveux central, le foie, les organes de reproduction de la femme et les glandes surrénales étaient les principaux organes cibles. Dans les études à long terme (26 et 39 semaines), la DSENO (dose sans effet nocif observable) pour les effets hépatiques, pour les effets sur la reproduction et pour les effets surrénaux était au moins quatre fois supérieure à l'exposition clinique sur la base de l'ASC.
Carcinogénicité et mutagénicité
Les essais de génotoxicité réalisés in vitro dans des systèmes bactériens et in vitro et in vivo dans des systèmes mammifères avec et sans activation métabolique n'indiquent pas de risque significatif chez l'Homme. Dans les études de carcinogénicité chez le rat et la souris, il a été observé une incidence accrue d'adénome/carcinome hépatocellulaire (à des doses plus faibles chez les hommes que chez les femmes) et des modifications néoplasiques d'adénome/carcinome folliculaire thyroïdien (chez le rat mâle uniquement). Les résultats sont probablement spécifiques aux rongeurs et considérés comme non pertinents pour l'Homme.
Fertilité et toxicité reproductive
Des études sur la reproduction chez le lapin et le rat ont montré une embryotoxicité, une foetotoxicité (des résorptions accrues et une viabilité foetale diminuée, une diminution du poids foetal, des malformations externes et des variations viscérales et squelettiques) et une tératogénicité à des doses toxiques pour la mère. La DSENO était égale à 10 fois l'exposition humaine (ASC) dans une étude de développement pré et post-natal et égale à 8 à 73 fois l'exposition humaine (ASC) dans une étude de fertilité et de développement embryonnaire précoce chez le rat.
Toxicité juvénile
Les résultats des études de toxicité chez les rats juvéniles concordaient largement avec ceux observés dans les études sur les rats adultes. Une maturation sexuelle retardée a été observée à des doses élevées sans effet sur la performance globale de reproduction ni sur les paramètres après une période de récupération de 6 semaines. Il n'y avait aucun effet sur la croissance des os longs ou sur le comportement.
Pas
d'exigences particulières.
Tout
médicament non utilisé ou déchet doit être éliminé conformément à la
réglementation en vigueur.
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en endocrinologie, diabète et maladies métaboliques ou en médecine interne.
Médicament nécessitant une surveillance particulière pendant le traitement
Comprimé pelliculé (comprimé).
Comprimés jaune pâle, ronds, biconvexes, à bords biseautés, portant l'inscription « Y1 » d'un côté et « NVR » de l'autre côté. Diamètre approximatif de 9,1 mm.
Flacon de 40 comprimés.